RevBayes Morphology Lab
Up to the Phylogenetics main page
Goals
This lab exercise will show how to analyze discrete morphological data using RevBayes. We will be using RevBayes v1.3.2 on the cluster in this lab.
This lab exercise is adapted from the tutorial entitled Discrete morphology - Tree Inference on the RevBayes web site.
Answer template
Here is a template in which to save your answers to the ![]() questions.
questions.
1. Is brlen_lambda a parameter or a hyperparameter in this model?
answer:
2. What is the posterior mean, standard deviation, and ESS (effective sample size) of tree length for the Mk model using the combined parameter samples from both runs?
answer:
posterior mean tree length:
standard deviation:
effective sample size:
3. How many total characters are in this data matrix?
answer:
4. How many characters in this data matrix are variable?
answer:
5. Do you expect estimated tree length to increase or decrease if we condition on variability? Briefly explain your reasoning.
answer:
6. What is the posterior mean, standard deviation, and ESS for tree length for the Mkv model using the combined parameter samples from both runs?
answer:
posterior mean tree length:
standard deviation:
effective sample size:
7. Did the Mkv model have the effect you expected?
answer:
8. In lecture, you were shown simulation results that suggested that estimates of edge lengths would be severely wrong unless the model conditioned on variability. Why did it make so little difference here?
answer:
9. What do the values in the categ_means vector represent? Note: do not say these are the category means, which you can deduce from the name of the variable. Instead, state how these values are used in the likelihood calculation.
answer:
10. What do the values in the categ_probs vector represent?
answer:
11. Of the 3 models, which fits substantially worse than the others?
answer:
12. Does allowing heterogeneity in state frequency provide substantially better fit than simply assuming equal state frequencies?
answer:
13. If you were preparing to publish a paper relating to these analyses, you probably would not want to state in your paper that you determined model fit by simply eye-balling trace plots. What quantity would you estimate to convince reviewers that one of these models fits the data worse than the other two?
answer:
Getting started
![]() Login to your account on the Storrs HPC cluster and start an interactive slurm session. If you have updated your
Login to your account on the Storrs HPC cluster and start an interactive slurm session. If you have updated your gensrun alias, you can just type:
ssh hpc
gensrun
Otherwise, type
ssh hpc
srun -p general -q general --mem=5G --pty bash
Important The --mem=5G part is important for this lab, as RevBayes sometimes uses more than the default amount of memory.
Create a directory
![]() Use the unix
Use the unix mkdir command to create a directory to play in today:
cd
mkdir morphlab
Important: be sure to not mis-spell the name of this directory when creating it, as much of the lab assumes that this directory exists!
Load module needed
![]() The RevBayes executable file needs some runtime libraries that can be loaded using the module system. Loading the R module loads the runtime library needed by rb132:
The RevBayes executable file needs some runtime libraries that can be loaded using the module system. Loading the R module loads the runtime library needed by rb132:
module load r/4.3.2
![]() You should now be able to type rb132 to start the program:
You should now be able to type rb132 to start the program:
RevBayes version (1.3.2)
Build from tags/v1.3.2 (rapture-4486-g3d84ac) on Sat Feb 28 11:57:57 EST 2026
Visit the website www.RevBayes.com for more information about RevBayes.
RevBayes is free software released under the GPL license, version 3. Type 'license()' for details.
To quit RevBayes type 'quit()' or 'q()'.
Download and save the data file
![]() Use the
Use the curl command (“Copy URL”) to download the data file for this tutorial from the RevBayes web site:
cd ~/morphlab
curl -O https://revbayes.github.io/tutorials/morph_tree/data/bears.nex
Creating a RevBayes script to explore the Mk model
Create a RevBayes script to carry out an MCMC analysis using the simplest 2-state Mk model. This script will be similar to the ones you created in previous labs, but this time we will add some additional variables to make it easier to modify the script later.
![]() Use nano to create a file (I’m assuming you are currently in the ~/morphlab folder where your data file is located.
Use nano to create a file (I’m assuming you are currently in the ~/morphlab folder where your data file is located.
nano mk.Rev
The scripts we will create today will end up being very similar to the files mcmc_mk.Rev, mcmc_mkv.Rev, and mcmc_mkv_discretized.Rev (written by April M. Wright, Michael J. Landis, and Sebastian Höhna) from the Discrete morphology tutorial.
Read the data file
![]() Enter the following text and then save the file:
Enter the following text and then save the file:
####################
# Useful variables #
####################
datafile <- "bears.nex"
logfile <- "output/mk.log"
treefile <- "output/mk.trees"
maptreefile <- "output/mk.map.tre"
majruletreefile <- "output/mk.majrule.tre"
nindepruns <- 1
nburnin <- 5000
ngen <- 50000
ntune <- 200
screenfreq <- 100
logfreq <- 10
treefreq <- 10
do_mcmc <- TRUE
do_summarize_trees <- FALSE
###############
# Data matrix #
###############
morpho <- readDiscreteCharacterData(datafile)
taxa <- morpho.names()
num_taxa <- taxa.size()
num_branches <- 2 * num_taxa - 2
moves = VectorMoves()
monitors = VectorMonitors()
Note that we’ve already used one of the variables we created at the beginning of the file (datafile).
Note also that we have specified nindepruns <- 1. Although each invokation of mk.Rev will perform just 1 MCMC analysis, we will (as you will see below) use a slurm script to perform 2 independent runs in parallel, so it will not take any longer to do 2 runs than it takes to do 1 run.
Add a tree model
The tree model specifies a prior for tree topology as well as a prior for edge lengths. We will also use this section to specify proposals (called moves in RevBayes) for modifying both tree topology and edge lengths.
![]() Add this text to your growing mk.Rev file:
Add this text to your growing mk.Rev file:
##############
# Tree model #
##############
brlen_lambda ~ dnExponential(0.1)
moves.append( mvScale(brlen_lambda, weight=2) )
phylogeny ~ dnUniformTopologyBranchLength(taxa, branchLengthDistribution=dnExponential(brlen_lambda))
tree_length := phylogeny.treeLength()
moves.append( mvNNI(phylogeny, weight=num_branches/2.0) )
moves.append( mvSPR(phylogeny, weight=num_branches/10.0) )
moves.append( mvBranchLengthScale(phylogeny, weight=num_branches) )
Is brlen_lambda a parameter or a hyperparameter in this model?
Create a substitution model
![]() Append the following to your script to specify a substitution model:
Append the following to your script to specify a substitution model:
######################
# Substitution Model #
######################
Q <- fnJC(2)
gamma_shape ~ dnExponential(0.01)
moves.append( mvScale(gamma_shape,lambda=1, weight=2.0) )
gamma_rate := gamma_shape
gamma_rates := fnDiscretizeGamma( gamma_shape, gamma_rate, 4 )
This tells RevBayes to create a Jukes-Cantor instantaneous rate matrix with 2 states (also known as a 2-state Mk, or M2, model) and store it in the variable Q.
The PhyloCTMC node puts everything together
![]() Append the following to your mk.Rev file:
Append the following to your mk.Rev file:
#############
# PhyloCTMC #
#############
# Specify the probability distribution of the data given the model
likelihood ~ dnPhyloCTMC(tree=phylogeny, siteRates=gamma_rates, Q=Q, type="Standard")
# Attach the data
likelihood.clamp(morpho)
mymodel = model(phylogeny)
Ready for MCMC
![]() We’ve now completely specified the model, so all that’s left is to create some monitors so that results are saved and set up the mcmc command:
We’ve now completely specified the model, so all that’s left is to create some monitors so that results are saved and set up the mcmc command:
#################
# MCMC Analysis #
#################
# Add monitors
monitors.append( mnModel(filename=logfile, printgen=logfreq) )
monitors.append( mnFile(filename=treefile, printgen=treefreq, phylogeny) )
monitors.append( mnScreen(printgen=screenfreq) )
# Start the MCMC analysis
if (do_mcmc) {
mymcmc = mcmc(mymodel, monitors, moves, nruns=nindepruns, combine="mixed")
mymcmc.burnin(generations=nburnin, tuningInterval=ntune)
mymcmc.run(generations=ngen)
# Check the performance of the moves (also known as operators)
mymcmc.operatorSummary()
}
if (do_summarize_trees) {
# Read in the tree trace and construct the consensus tree
trace = readTreeTrace(treefile, treetype="non-clock")
trace.setBurnin(0.25)
# Summarize tree trace and the consensus tree to file
mapTree(trace, file=maptreefile)
consensusTree(trace, file=majruletreefile)
}
# Quit RevBayes
quit()
Note that we used the boolean (yes/no) variables do_mcmc to determine whether to perform the MCMC analysis and do_summarize_trees to determine whether the tree file will be processed to create consensus and a MAP (maximum a posteriori) tree. Currently we have set do_mcmc <- TRUE and do_summarize_trees <- FALSE, so an MCMC analysis will be performed but no MAP or consensus tree will be generated. This is prudent because summarizing the tree file takes a lot of time and memory and is unnecessary because we will actually not even look at the MAP or consensus trees during this lab. If you, later, want to see the MAP and consensus trees from this run, you can re-run mk.Rev after setting do_mcmc <- FALSE and do_summarize_trees <- TRUE.
Create a slurm script
We will use sbatch in this lab to perform our runs, so we will need to create a slurm script.
![]() Use nano to create a file named mk.slurm with the following contents:
Use nano to create a file named mk.slurm with the following contents:
#!/bin/bash
#SBATCH --job-name=mk # Job name
#SBATCH --output=mk%a.out # stdout: %a becomes the array id
#SBATCH --error=mk%a.err # stderr: %a becomes the array id
#SBATCH --array=1-2 # Perform 2 independent runs in parallel
#SBATCH --partition=general # Name of Partition
#SBATCH --ntasks=1 # Maximum CPU cores for each job
#SBATCH --nodes=1 # Ensure all cores are from the same node for each job
#SBATCH --cpus-per-task=1 # CPU-cores per task for each job
#SBATCH --constraint='epyc128' # Target the AMD Epyc 128-core node architecture
#SBATCH --mem=5G # Request 20GB of available RAM
# Create environmental variable telling R where to
# find your locally-installed convenience package
export R_LIBS="$HOME/rlib"
# Load R for convergence analysis (also provides run-time library needed by RevBayes)
module load r/4.3.2
# Remove previous results if there are any
rm -rf ~/morphlab/mkrun$SLURM_ARRAY_TASK_ID
# Create a directory in which to work and navigate into it
mkdir -p ~/morphlab/mkrun$SLURM_ARRAY_TASK_ID
cd ~/morphlab/mkrun$SLURM_ARRAY_TASK_ID
# Copy the data file into the directory just created
# so that RevBayes can find it
cp ../bears.nex .
# Run RevBayes
rb132 ../mk.Rev
The $SLURM_ARRAY_TASK_ID will be replaced with the job number. We specified two jobs (#SBATCH --array=1-2) numbered 1 and 2, so two directories will be created by the mkdir command, _ ~/morphlab/mkrun1_ and ~/morphlab/mkrun2. The way to think about this is that the commands following the #SBATCH directives in _mk.slurm will be executed twice: the first time $SLURM_ARRAY_TASK_ID will be set to 1, and the second time $SLURM_ARRAY_TASK_ID will be set to 2.
Run the slurm script
![]() Type the following to start your run:
Type the following to start your run:
sbatch mk.slurm
All you will see is a line like this (the job id will differ of course):
Submitted batch job 23288390
![]() Check the progress of your run periodically using the
Check the progress of your run periodically using the squeue command and specifying your NetID (not the one shown below, which is Paul’s NetID!):
squeue -u pol02003
If your jobs are still running, you will see something like this:
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
23289185_2 general mkv pol02003 R 0:09 1 cn504
23289185_1 general mkv pol02003 R 0:10 1 cn468
23288505 general bash pol02003 R 47:47 1 cn502
The jobs with id 23289185_1and 23289185_2 are my MCMC analyses, while the job with id 23288505 is my interactive session.
If your jobs have finished (or if there was an error), you will see just the interactive session:
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
23288505 general bash pol02003 R 53:59 1 cn502
if you discover you’ve done something wrong and want to cancel the jobs you’ve started, use the scancel command followed by the job id that you got from running squeue:
scancel 23289185
Note that 23289185 is my job id (you should not specify this one!) and note also that specifying 23289185 cancels both 23289185_1 and 23289185_2.
Check convergence
![]() While your array job is running, create a file convergence.R inside your ~/morphlab directory using nano and insert this text:
While your array job is running, create a file convergence.R inside your ~/morphlab directory using nano and insert this text:
.libPaths(c("~/rlib", .libPaths()))
library(convenience)
checkConvergence(list_files=c("mkrun1/output/mk.log","mkrun1/output/mk.trees","mkrun2/output/mk.log","mkrun2/output/mk.trees"))
![]() Once your array job is finished, run convergence.R in R to check convergence of your two independent runs:
Once your array job is finished, run convergence.R in R to check convergence of your two independent runs:
Rscript convergence.R
Viewing the log files in Tracer
Note that RevBayes saved the output in two directories named mkrun1/output and mkrun2/output, which were generated because you included output/ in each of the output file paths in your mk.Rev script.
![]() Download the files mkrun1/output/mk.log and mkrun2/output/mk.log to your local laptop and open them in Tracer and, after selecting both files in the Trace Files section, click the Marginal Density tab and look through density plots for the
Download the files mkrun1/output/mk.log and mkrun2/output/mk.log to your local laptop and open them in Tracer and, after selecting both files in the Trace Files section, click the Marginal Density tab and look through density plots for the Posterior, Likelihood, Prior, brlen_lambda, gamma_shape, and tree_length. The two independent runs should have yielded very similar results.
![]() Now click on “Combined” in Tracer’s “Trace Files” list, click the “Estimates” tab at the top, and answer the following question:
Now click on “Combined” in Tracer’s “Trace Files” list, click the “Estimates” tab at the top, and answer the following question:
Conditioning on variability using the Mkv model
We will now see what effect conditioning on variability makes. When the Mk model conditions on variability, it is denoted the Mkv model.
![]() Use the exclude (exclude character) command in PAUP* to determine how many variable characters are present in the data set we are using:
Use the exclude (exclude character) command in PAUP* to determine how many variable characters are present in the data set we are using:
paup bears.nex
paup> exclude constant
paup> quit
Perform a second MCMC analysis using the Mkv model
![]() Start by copying your mk.Rev script, naming the copy mkv.Rev:
Start by copying your mk.Rev script, naming the copy mkv.Rev:
cp mk.Rev mkv.Rev
![]() Also copy your mk.slurm script, naming the copy mkv.slurm:
Also copy your mk.slurm script, naming the copy mkv.slurm:
cp mk.slurm mkv.slurm
![]() Finally, copy your convergence.R script, naming the copy convergence-mkv.R:
Finally, copy your convergence.R script, naming the copy convergence-mkv.R:
cp convergence.R convergence-mkv.R
![]() Now open mkv.Rev in nano and change all of these lines:
Now open mkv.Rev in nano and change all of these lines:
logfile <- "output/mk.log"
treefile <- "output/mk.trees"
maptreefile <- "output/mk.map.tre"
majruletreefile <- "output/mk.majrule.tre"
to these:
logfile <- "output/mkv.log"
treefile <- "output/mkv.trees"
maptreefile <- "output/mkv.map.tre"
majruletreefile <- "output/mkv.majrule.tre"
Note how easy is is to make these changes since all the file names are stored in variables near the top of the file!
![]() The only change we need to make to cause RevBayes to condition on variability is to add coding=”variable” to our PhyloCTMC call:
The only change we need to make to cause RevBayes to condition on variability is to add coding=”variable” to our PhyloCTMC call:
#############
# PhyloCTMC #
#############
# Specify the probability distribution of the data given the model
likelihood ~ dnPhyloCTMC(tree=phylogeny, siteRates=gamma_rates, Q=Q, type="Standard", coding="variable")
![]() Open mkv.slurm in nano and change
Open mkv.slurm in nano and change mk to mkv in 7 places. You can easily find these places using Ctrl-W and typing in what you are searching for (i.e. mk). Be careful not to change mkdir to mkvdir, however!
![]() Rerun RevBayes using the mkv.slurm script:
Rerun RevBayes using the mkv.slurm script:
sbatch mkv.slurm
![]() While the run is going, edit your convergence-mkv.R script so that you will be ready to test convergence.
While the run is going, edit your convergence-mkv.R script so that you will be ready to test convergence.
![]() Once your runs finish, run convergence-mkv.R to test convergence:
Once your runs finish, run convergence-mkv.R to test convergence:
Rscript convergence-mkv.R
This time you will find that the runs were not long enough to achieve convergence. Given the time available for this lab, we will not try to fix this, but if you were doing this analysis for publication, you should ensure that the results you are reporting are from an MCMC analysis that has achieved convergence.
![]() Download (to your local laptop) the file mkvrun1/output/mkv.log and mkvrun2/output/mkv.log file into Tracer
Download (to your local laptop) the file mkvrun1/output/mkv.log and mkvrun2/output/mkv.log file into Tracer
Allowing Beta-distributed state frequencies
We will try one more modification to our Mk model today. The Mk and Mkv models we’ve used thus far all assume that the forward substitution rate is equal to the reverse substitution rate. This leads to equal equilibrium state frequencies (i.e. \(\pi_0 = \pi_1 = 0.5\)).
We can relax this (as described in lecture) by allowing the likelihood to be a mixture model, thus providing several choices of \(\pi_0\) (and thus \(\pi_1\)) for each character. Some characters will end up giving more weight to smaller values of \(\pi_0\) while other characters will end up giving more weight to larger values.
There are 459 0s and 466 1s in the data matrix, so the overall proportions of 0s and 1s are nearly equal; however, the fraction of 0s for any given character ranges from 0.1 to 0.8, so there is room to allow the frequencies of state 0 and state 1 to vary across characters.
![]() Copy your mkv.Rev script, naming the copy mkvbeta.Rev:
Copy your mkv.Rev script, naming the copy mkvbeta.Rev:
cp mkv.Rev mkvbeta.Rev
![]() Copy your mkv.slurm script, naming the copy mkvbeta.slurm:
Copy your mkv.slurm script, naming the copy mkvbeta.slurm:
cp mkv.slurm mkvbeta.slurm
![]() Copy your convergence-mkv.R script, naming the copy convergence-mkvbeta.R:
Copy your convergence-mkv.R script, naming the copy convergence-mkvbeta.R:
cp convergence-mkv.R convergence-mkvbeta.R
Modifying your RevBayes script
![]() Now open mkvbeta.Rev in nano and change all of these lines:
Now open mkvbeta.Rev in nano and change all of these lines:
logfile <- "output/mkv.log"
treefile <- "output/mkv.trees"
maptreefile <- "output/mkv.map.tre"
majruletreefile <- "output/mkv.majrule.tre"
to this:
logfile <- "output/mkvbeta.log"
treefile <- "output/mkvbeta.trees"
maptreefile <- "output/mkvbeta.map.tre"
majruletreefile <- "output/mkvbeta.majrule.tre"
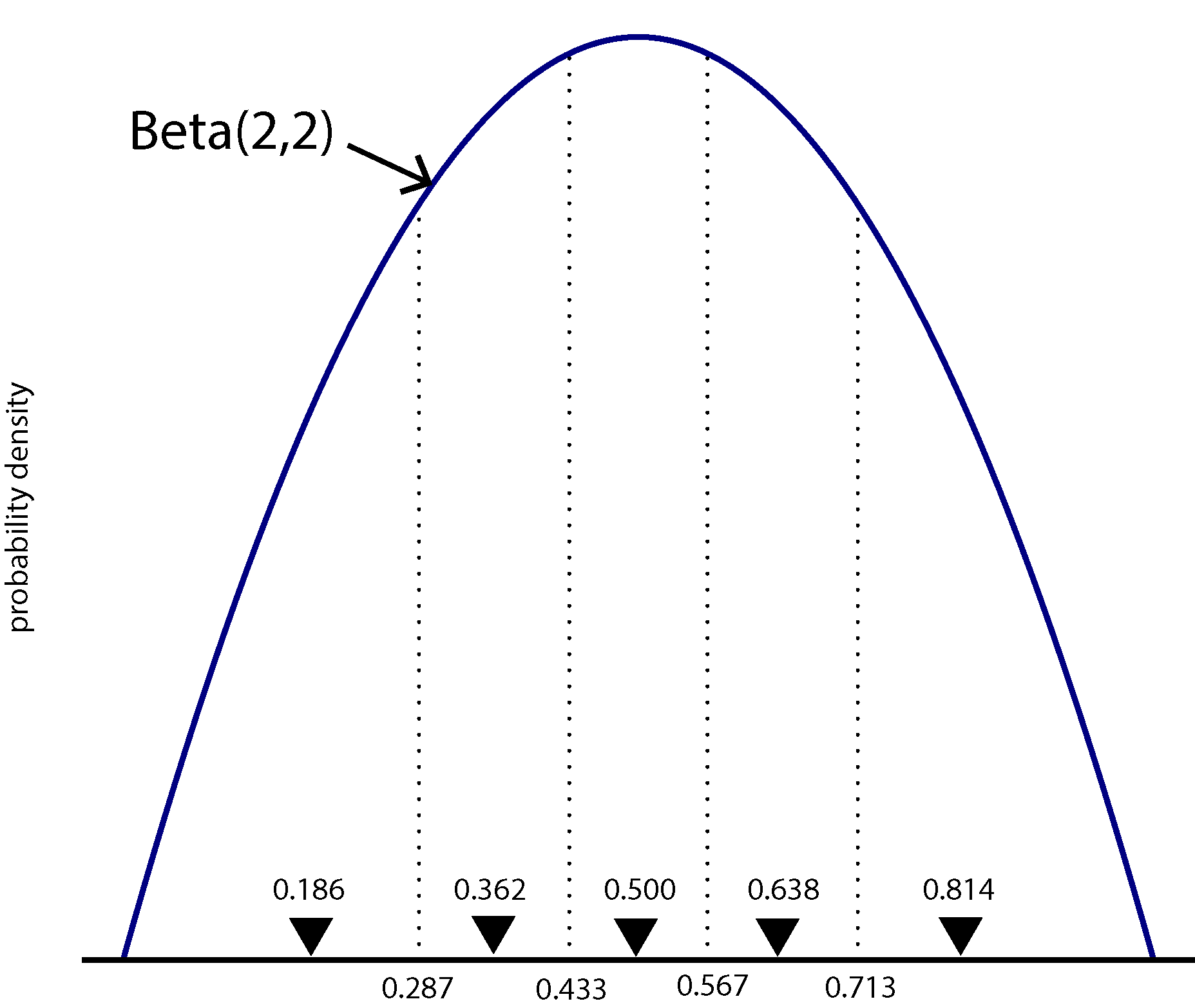
We will model among-character frequency variation in much the same way we model among-character rate variation, using a discretized probability distribution. The main difference is that frequencies are constrained to the part of the real number line between 0.0 and 1.0, so we will use a discrete Beta distribution rather than the discrete Gamma distribution used for modeling rate heterogeneity.
The figure above shows a Beta(2,2) density divided into 5 equal-area categories, with the mean of each category shown using a downward-pointing filled triangle. The likelihood for one character will be calculated using 5 different rate matrices, each using \(\pi_0\) from a different category mean. Each component of the mixture will have probability 0.2, as that is the area under the Beta density curve for each category (assuming there are 5 categories).
Thus, we need 5 different Q matrices in our model, one for each category, so Q in our script will now be a vector of rate matrices rather than a single rate matrix. The shape parameter beta_shape used to determine the symmetric Beta(beta_shape,beta_shape) distribution used will be allowed to vary during the MCMC run, so the 5 Q matrices must be recalculated each time beta_shape changes. This is accomplished by making each element of the Q vector a deterministic node. The recalculation of each rate matrix is accomplished by the fnDiscretizeBeta function (just as the fnDiscretizeGamma function handles recalculation of relative rates for the discrete gamma rate heterogeneity model as the gamma_shape parameter changes).
![]() Replace the entire Substitution Model section in mkvbeta.Rev. Everything here has changed except for the last 5 lines, and all but the last are related to rate heterogeneity and are the same lines we used for the Mkv model.
Replace the entire Substitution Model section in mkvbeta.Rev. Everything here has changed except for the last 5 lines, and all but the last are related to rate heterogeneity and are the same lines we used for the Mkv model.
######################
# Substitution Model #
######################
# Specify number of categories to create
num_beta_cats = 5
# Create the parameter that will determine the shape of the Beta distribution
beta_shape ~ dnLognormal( 1.0, sd=1.0 )
moves.append( mvScale(beta_shape, lambda=1, weight=5.0 ) )
# Calculate means of each category
beta_shape.setValue(2.0);
categ_means := fnDiscretizeBeta(beta_shape, beta_shape, num_beta_cats)
print("categ_means = ",categ_means)
# Create vector of Q matrices
for (i in 1:num_beta_cats) {
Q[i] := fnF81(simplex(categ_means[i], abs(1-categ_means[i])))
}
categ_probs <- simplex( rep(1,num_beta_cats) )
print("categ_probs = ",categ_probs)
gamma_shape ~ dnExponential(0.01)
moves.append( mvScale(gamma_shape,lambda=1, weight=2.0) )
gamma_rate := gamma_shape
gamma_rates := fnDiscretizeGamma( gamma_shape, gamma_rate, 4 )
quit()
Before we continue, let’s run this script up to this point so that we can see the output from the two print statements in the code above. We want to stop processing the script right after this block of code, so I’ve inserted a quit() command just after the line that calls fnDiscretizeGamma:
rb132 mkvbeta.Rev
You can see that we are dividing the Beta distribution into 5 equal chunks (num_beta_cats = 5), providing the beta_shape parameter with a Lognormal prior distribution that heavily favors shapes greater than 1 (84%), which is good because shapes less than 1 create U-shaped Beta density functions that place most weight on the extreme values 0 and 1.
categ_meansvector represent? Note: do not say these are the category means, which you can deduce from the name of the variable. Instead, state how these values are used in the likelihood calculation.
Hint: compare them to the figure above. I’ve used the setValue function to ensure that the beta_shape parameter is initially equal to 2 (to correspond with the figure).
categ_probsvector represent?
We need to do one last thing in order for RevBayes to actually use the above substitution model. We need to inform the PhyloCTMC object that we are using a mixture distribution to model the Q matrix.
![]() To do this, add
To do this, add siteMatrices=categ_probs to the end of your dnPhyloCTMC function call:
likelihood ~ dnPhyloCTMC(tree=phylogeny, siteRates=gamma_rates, Q=Q, type="Standard", coding="variable", siteMatrices=categ_probs)
Modifying your slurm script and running the analysis
![]() Remove the quit() statement at the end of your Substitution Model section, modify mkv.slurm (changing all the
Remove the quit() statement at the end of your Substitution Model section, modify mkv.slurm (changing all the mkv parts to mkvbeta), and run the MCMC analyses:
sbatch mkvbeta.slurm
This will take some time because now RevBayes must calculate each site likelihood 5 times, once for each Q matrix in the mixture distribution.
Test for convergence
![]() While it is running, use the time to modify convergence-mkvbeta.R. Once both runs finish, test for convergence using:
While it is running, use the time to modify convergence-mkvbeta.R. Once both runs finish, test for convergence using:
Rscript convergence-mkvbeta.R
Perhaps surprisingly, for me at least, the test said that convergence had happened, despite the fact that this model is the most complex of the tree. You may or may not have the same outcome depending on the variation in your runs compared to mine.
Comparing Mk, Mkv, and the Mkv beta mixture model
![]() Load mkrun1/output/mk.log, mkvrun1/output/mkv.log, and mkvbetarun1/output/mkvbeta.log into Tracer and compare their likelihoods using the Estimates and/or Marginal Density tab. Note that you will need to select all three files in the Trace Files section so that you can compare them side-by-side.
Load mkrun1/output/mk.log, mkvrun1/output/mkv.log, and mkvbetarun1/output/mkvbeta.log into Tracer and compare their likelihoods using the Estimates and/or Marginal Density tab. Note that you will need to select all three files in the Trace Files section so that you can compare them side-by-side.
What to turn in
Turn in your answers to the ![]() thinking questions on the template provided above. Send them to Analisa via Slack.
thinking questions on the template provided above. Send them to Analisa via Slack.