Likelihood Lab
Up to the Phylogenetics main page
Goals
The goal of this lab exercise is to show you how to conduct maximum likelihood analyses in PAUP* using several models, and to decide among competing models using likelihood ratio tests.
Getting started
Login to your account on the Storrs HPC cluster:
ssh hpc
Type the following:
srun -p general -q general --pty bash
If you get tired of typing all this, you can create an alias by opening the .bashrc file in your home directory:
nano ~/.bashrc
and typing the following (you can make it the first line of the file):
alias gensrun='srun -p general -q general --pty bash'
Save the .bashrc file and you can simply type gensrun to run the srun command the next time you login to the cluster!
Create a text file in which to save answers
You will be submitting your answers to the questions posed in the boxes labeled with the ![]() emoji, so create a text file on your local computer using either BBEdit (if you are a Mac user) or Notepad++ (if you are a Windows user) and paste the text below into the new text file. Please place your answer below the line containing the question, and separate your answer from the next question by a blank line to make it easier for us to review.
emoji, so create a text file on your local computer using either BBEdit (if you are a Mac user) or Notepad++ (if you are a Windows user) and paste the text below into the new text file. Please place your answer below the line containing the question, and separate your answer from the next question by a blank line to make it easier for us to review.
Are chlorophyll-b taxa together in the F81 tree?
answer:
What are the empirical base frequencies for this data set?
answer:
What is the log likelihood of this tree under this "empirical base frequencies" version of the F81 model?
answer:
What proportion of sites are constant?
answer:
What are the maximum likelihood estimates (MLEs) of the base frequencies?
answer:
What is the log likelihood of this tree under the "estimated base frequencies" version of the F81 model?
answer:
What parameters are being estimated using the F81 model?
answer:
Does this model fit the data better than the "empirical base frequencies" version of the F81 model? What information did you use to come to your conclusion?
answer:
What is the MLE of the transition/transversion ratio under the HKY85 model?
answer:
What is the MLE of the transition/transversion _rate_ ratio under the HKY85 model?
answer:
What is the log likelihood of this tree under the HKY85 model?
answer:
What parameters are being estimated using the HKY85 model?
answer:
Does the HKY model fit the data better than the F81 model?
answer:
The transition/transversion rate ratio (kappa) is the ___ divided by the ___.
answer:
The transition/transversion ratio (tratio) is the ___ divided by the ___.
answer:
What is the MLE of pinvar under the HKY85+I model?
answer:
Is the MLE of pinvar larger or smaller than the proportion of constant sites?
answer:
Why are these two proportions different? That is, how can a site be constant but not invariable?
answer:
What is the log likelihood of this tree under the HKY85+I model?
answer:
What parameters are being estimated using the HKY85+I model?
answer:
What is the MLE of the gamma shape parameter under the HKY85+G model?
answer:
What is the log likelihood of this tree under the HKY85+G model?
answer:
What parameters are being estimated using the HKY85+G model?
answer:
What is the MLE of the gamma shape parameter under the HKY85+I+G model?
answer:
What is the MLE of the pinvar parameter under the HKY85+I+G model?
answer:
Is the MLE of the shape parameter higher or lower under the HKY85+I+G model compared to the HKY85+G model? Explain why this is so.
answer:
What is the log likelihood of this tree under the HKY85+I+G model?
answer:
What parameters are being estimated using the HKY85+I+G model?
answer:
What is the likelihood ratio test statistic for F81 vs. HKY85?
answer:
How many degrees of freedom for this test?
answer:
What is the significance (P-value) for this test?
answer:
Does allowing for a transition/transversion bias make a significant difference?
answer:
What is the likelihood ratio test statistic for a comparison of HKY+I to HKY?
answer:
What is the critical value for this likelihood ratio test (of HKY vs HKY+I)? That is, what is the smallest likelihood ratio test statistic that would be significant at the 0.05 level?
answer:
Does the HKY85+I model explain the data significantly better than an equal rates HKY85 model?
answer:
Does the HKY85+G model explain the data significantly better than an equal rates HKY85 model?
answer:
Does the HKY85+G model explain the data better than HKY85+I? Why can't you use a likelihood ratio test to compare these two models?
answer:
Does the HKY85+I+G model explain the data significantly better than either HKY85+I or HKY85+G alone?
answer:
Does the model you have selected place all the chlorophyll-b organisms together?
answer:
Which model (of the 5 you tested) is best according to AICc?
answer:
Which model (of the 5 you tested) is best according to BIC?
answer:
Create a directory for this lab
Create a new directory to use for this lab as follows:
cd
mkdir likelab
cd likelab
(The first cd all by itself is just to ensure that you are in your home directory before you create the new likelab directory.) Creating a directory just for this lab will avoid overwriting (or accidentally using) files from previous labs.
Download the data file algae.nex
Download the data file algae.nex using the following curl command on the cluster:
curl -Ok https://gnetum.eeb.uconn.edu/courses/phylogenetics/lab/algae.nex
If you remember from lecture, adding more parameters to a model to account for different aspects of nucleotide and sequence evolution can – but does not necessarily – improve the explanatory ability of a model, or its ability to produce a correct phylogeny. Our goal for this lab will be to see if we can tease apart which aspects of sequence evolution are most important for getting the tree correct.
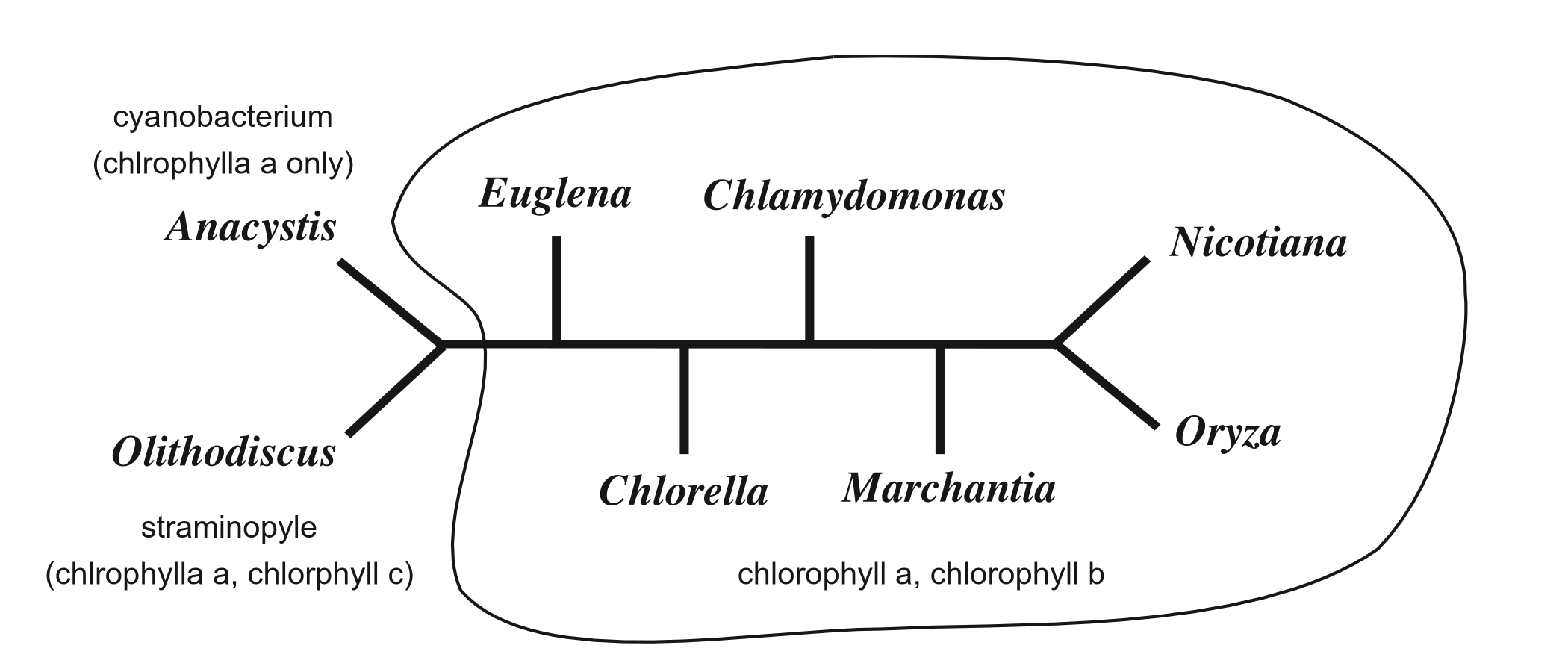
The accepted phylogeny (based on much evidence besides these data) places all the chlorophyll-b-containing plastids together (Lockhart et al. 1994).
Thus, there should be a branch in the tree separating all chlorophyll-b-containing taxa (circled in the figure above) from the two taxa that do not have chlorophyll b, namely the cyanobacterium Anacystis (which has chlorophyll a and phycobilin accessory pigments) and the straminopyle Olithodiscus (which has chlorophylls a and c).
Obtain the maximum likelihood tree under the F81 model
The first goal is to learn how to obtain maximum likelihood estimates of the parameters in several different substitution models. Use PAUP* to answer the following questions. Start by obtaining the maximum likelihood tree under the F81 model. Create a run.nex file and save it with the following contents:
#nexus
begin paup;
execute algae.nex;
set criterion=likelihood;
lset nst=1 basefreq=empirical;
hsearch;
end;
The nst=1 tells PAUP* that we want a model having just one substitution rate parameter (the JC69 and F81 models both fall in this category). The basefreq=empirical tells PAUP* that we want to use simple estimates of the base frequencies. The empirical frequency of the base G, for example, is the value you would get if you simply counted up all the Gs in your entire data matrix and divided by the total number of nucleotides. The empirical frequencies are not usually the same as the maximum likelihood estimates (MLEs) of the base frequencies, but they are quick to calculate and often very close to the corresponding MLEs.
Execute run.nex in PAUP* (i.e. type paup run.nex on the command line). Because there is no quit command in the paup block, PAUP* will stay around, produce a paup> prompt, and wait for you to type a command. Issue the following command to show the tree:
showtrees;
One problem is that the tree is drawn in such a way that it appears to be rooted within the flowering plants (tobacco and rice). Specifying the cyanobacterium Anacystis as the outgroup makes more sense biologically. To set this, type the following at the paup> prompt:
outgroup Anacystis_nidulans;
showtrees;
You could also have used this command:
outgroup 7;
showtrees;
because Anacystis is the 7th (of 8) taxa in the data matrix.
The edge lengths are not drawn proportional to the expected number of substitutions when using the showtrees command. To fix this, use the describetrees command rather than the simpler showtrees command:
descr 1 / plot=phylogram;
As with all PAUP* commands, it is usually not necessary to type the entire command name, only enough letters that PAUP* can determine unambiguously which command you want. Here, you typed descr instead of describetrees, and it worked just fine.
Are chlorophyll-b taxa together in the F81 tree?
You will be working with this tree for quite awhile. Resist the temptation to do heuristic searches with each model, as it will be important to compare the performance of all of the models on the same tree topology! To be safe, save this tree to a file named f81.tre using the savetrees command:
savetrees file=f81.tre brlens;
If you ever need to read this tree back in, use the gettrees command:
gettrees file=f81.tre;
Now get PAUP* to show you the maximum likelihood estimates for the parameters of the F81 model used in this analysis (the 1 here refers to tree 1 in memory):
lscores 1;
cstatuscommand will give you this information)
Build paup block as you go
I would like to recommend that, instead of typing commands at the paup> prompt, you instead (from now on) edit run.nex and add the new commands to your paup block. Here is a revised run.nex file with a paup block updated to include what we’ve already done:
#nexus
begin paup;
execute algae.nex;
set criterion=likelihood;
lset nst=1 basefreq=empirical;
[
hsearch;
outgroup Anacystis_nidulans;
showtrees;
descr 1 / plot=phylogram;
savetrees file=f81.tre brlens;
]
gettrees file=f81.tre;
lscores 1;
[add new commands here]
quit;
end;
(Be sure to type q to exit PAUP* before typing nano run.nex to edit this file.)
You’ll notice that I’ve commented out [using square brackets] everything from the hsearch command through saving the tree file. We need not do the search over again every time we run this file because the tree and branch lengths resulting from this search are already saved in the file f81.tre.
You will also notice that I added a quit command at the end. This causes PAUP* to quit after executing all the commands in the paup block, saving you the trouble of typing quit in order to edit the run.nex file in preparation for the next step.
This method (building up a paup block) has the advantage that you always have a document that provides a record of everything you’ve done. It is very easy to perform an analysis and not be able to repeat it later because you’ve forgotten some setting you changed along the way.
Estimate base frequencies
Now estimate the base frequencies on this tree with maximum likelihood by adding the following 3 lines to your paup block just above the quit command (you can replace the [add new commands here] comment). Note how the lscores command is used to force PAUP* to recompute the likelihood (under the revised model) and spit out the parameter estimates.
[F81 model with estimated base frequencies]
lset basefreq=estimate;
lscores 1;
Note: the new lset command will reset the previous one (which specified basefreq=empirical). This will allow you to see the difference in likelihood when using empirical vs estimated base frequencies. The first lscores 1 command will use empirical frequencies and the second lscores 1 command will use estimated frequencies. You may wish to make the comment [F81 model with estimated base frequencies] into a printed comment so that you have some explanation in the output describing what base frequency setting is being used.
Estimate transition/transversion bias
Switch to the HKY85 model now and estimate the transition/transversion ratio along with the base frequencies. The way you specify the HKY model in PAUP* is to tell it you want a model with 2 substitution rate parameters (nst=2), and that you want to estimate the base frequencies (basefreq=estimate) and the transition/transversion ratio (tratio=estimated). Note that these specifications also apply to the F84 model, so if you wanted PAUP* to use the F84 model, you would need to add variant=f84 to the lset command.
[F81 model with estimated base frequencies]
[lset basefreq=estimate;]
[lscores 1;]
[HKY85 model]
lset nst=2 basefreq=estimate tratio=estimate;
lscores 1;
(Note that I commented out the code for the F81 model.)
_______divided by the_______.
_______divided by the_______.
Estimate the proportion of invariable sites
Now ask PAUP* to estimate pinvar, the proportion of invariable sites, by adding pinvar=estimate to your existing lset command. The HKY85 model with among-site rate heterogeneity modeled using the two-category invariable sites approach is called the HKY85+I model.
Estimate the heterogeneity in rates among sites
Now change pinvar=estimate to pinvar=0 in your lset command and tell PAUP* to use the discrete gamma distribution with 5 rate categories. Here are the commands for doing this all in one step:
lset nst=2 basefreq=estimate tratio=estimate pinvar=0 rates=gamma ncat=5 shape=estimate;
lscores 1;
The HKY85 model with among-site rate heterogeneity modeled using the discrete gamma approach is called the HKY85+G model.
Estimate both pinvar and the gamma shape parameter
Now ask PAUP* to estimate both pinvar and the gamma shape parameter simultaneously (the HKY85+I+G model).
Likelihood ratio tests
In this section, you will perform some simple likelihood ratio tests to decide which of the models used in the previous section does the best job of explaining the data while keeping the number of parameters used to a minimum.
Determining significance
A model having k parameters can always attain a higher likelihood than any model having fewer than k parameters that is nested within it (you should be able to explain why this is true), so the question we will be asking is whether more complex (i.e. more parameter-rich) models fit significantly better than simpler nested models. To do this we will assume that the likelihood ratio test statistic LR (equal to twice the difference in log-likelihoods) has the same distribution as a chi-squared random variable with degrees of freedom (d.f.) equal to the difference in the number of estimated parameters in the two models. (A parameter whose value is fixed or which can be determined from the values of other parameters doesn’t count as an estimated parameter.)
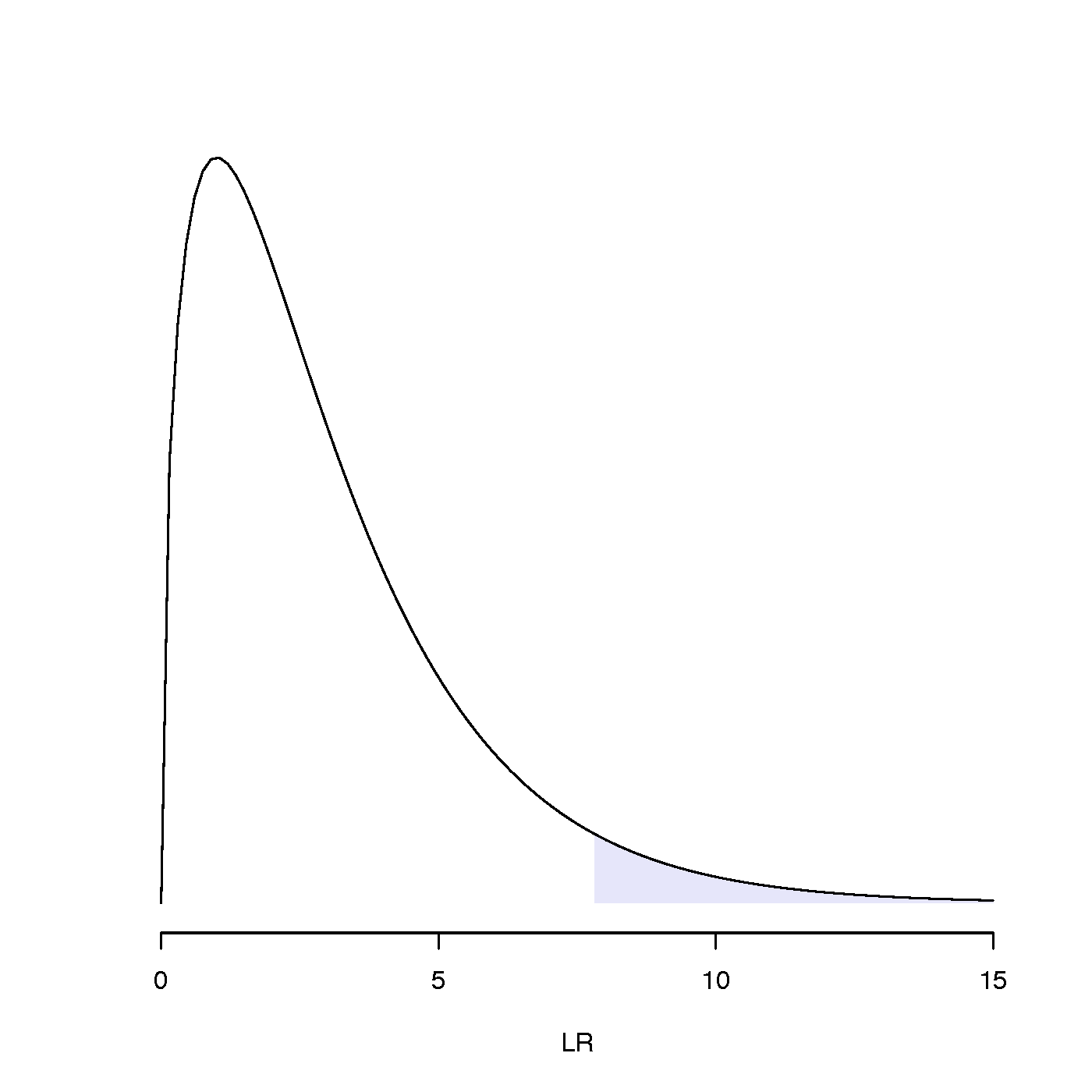
To be specific, we would like to know whether the LR test statistic falls inside the 5% right tail of the chi-squared distribution (see figure to the right for an example). If it does, then it should be considered an unusually large value of LR; i.e. not a LR value that would normally arise (i.e. falls within the interval that accounts for 95% of the probability distribution) if the models were equivalent explanations of the data.
We can use R to do the calculation for us. R is available as a module on the cluster. To load this module, be sure you have exited PAUP* and type:
module load r
You should see a response like this:
Loading r/4.5.2
Loading requirement: gcc/14.2.0
Now type R to start R:
R
Suppose LR = 6.91 (twice the difference in log likelihood between the models, which is 3.455) and d.f. = 1 (one parameter differs between the models). To ask R to tell us what fraction of the chi-square distribution (with 1 degree of freedom) is to the left of 6.91, use the pchisq (chi-squared cumulative probability) command:
pchisq(6.91, df=1)
You should get this response
[1] 0.9914285
which tells us that 99.14285% of the distribution is to the left of 6.91 and thus less than 1% is to the right, which means 6.91 is significantly large (because the probability of seeing a value that large or larger is less than 5%).
To find the critical value, you can use the qchisq (chi-squared quantile) command:
qchisq(0.95, df=1)
This tells us the specific value that we have to exceed in order to be significant. In this case (when d.f.=1), it is 3.841459.
To escape R, type q() and then answer n when asked if you want to save the workspace image.
What parameters make the fit of the model significantly better?
The model we will begin with is the F81 model with estimated base frequencies. Compare this F81 model to the HKY85 model, which differs from the F81 model only in the fact that it allows transitions and transversions to occur at different rates.
To calculate the likelihood ratio test statistic LR, subtract the log-likelihood of the less complex model from that of the more complex model and multiply by 2. This will give you a positive number. If you ever get a negative LR test statistic, it means you have the models in the wrong order.
You should have all the numbers you need to perform these likelihood ratio tests. If, however, you have not written some of them down, and thus need to redo some of these analyses, you might need to know how to turn off rate heterogeneity using the following command:
lset rates=equal pinvar=0;
It will help if you create a table that lists the (maximized) log-likelihood and number of free parameters for the models F81, HKY, HKY+I, HKY+G, and HKY+I+G before you start trying to answer the questions below.
Repeat search under a better model
Earlier in the lab, you found that the F81 model with empirical base frequencies did not produce the expected tree separating chlorophyll-b organisms from the two (Anacystis and Olithodiscus) that lack chlorophyll-b. Would a new search under a better model have the same result?
Using the simplest model that you can defend (of the five we have examined: F81, HKY85, HKY85+I, HKY85+G, HKY85+I+G), perform a heuristic search under the maximum likelihood criterion. Use the template below, but make changes necessary to match the model you have chosen to use.
The next two paragraphs provide some explanation of the commands used in the template.
To make the analysis go faster, we will ask PAUP* to not re-estimate all the model parameters for every tree it examines during the search. To do this, first use the lset command to set up the model you are planning to use (be sure to specify estimate for all relevant parameters: base frequencies, shape, tratio, etc.). Use the lscores command to force PAUP* to re-estimate all of the parameters of your selected model on some tree (the tree just needs to be something reasonable, such as a NJ tree or the F81 tree you have been using).
Now, re-issue the lset command but, for every parameter that you estimated, change the word estimate to previous. After executing this new lset command, start a search using just hsearch. PAUP* will fix the parameters at the previous values (i.e. the estimates you just forced it to calculate) and use these same values for every tree examined during the search.
Template Supposing you decided that HKY85+I+G was the best model, here is how your run.nex file should be set up:
#nexus
begin paup;
execute algae.nex;
set criterion=likelihood;
[Estimate HKY85+I+G parameters on a neighbor-joining tree]
lset nst=2 basefreq=estimate tratio=estimate rates=gamma ncat=5 shape=estimate pinvar=estimate;
nj;
lscores 1;
[Fix parameters at their estimated values]
lset nst=2 basefreq=previous tratio=previous rates=gamma ncat=5 shape=previous pinvar=previous;
[Perform heuristic search under the HKY85+I+G model]
hsearch;
[Show and save resulting tree]
outgroup Anacystis_nidulans;
showtrees;
describe 1 / plot=phylogram;
savetrees file=hky85ig.tre brlens replace;
quit;
end;
Automodel
You might be exhausted by now, but let’s try one last (very useful!) command.
Use nano to create a new “run” file named automodel.nex:
nano automodel.nex
Copy and paste the following text into this new file:
#nexus
begin paup;
[Start a log file]
log start file=pauplog.txt replace;
[Read the data]
execute algae.nex;
[Set up for likelihood]
set criterion=likelihood;
[Read in the tree you've been using for model testing]
outgroup Anacystis_nidulans;
gettrees file=f81.tre;
[Test all models at once!]
automodel;
[Stop logging to pauplog.txt and quit]
log stop;
quit;
end;
Run this file in PAUP*:
paup automodel.nex
This generates a lot of output, but it is easy to pick out the few models you tested and check to ensure that you got the number of parameters and log-likelihood correct when you did things the hard way. This table also evaluates models using AICc and BIC.
Literature Cited
Lockhart, P. J., Steel, M. A., Hendy, M. D., & Penny, D. 1994. Recovering evolutionary trees under a more realistic model of sequence evolution. Molecular Biology and Evolution 11(4):605–612.