IQ-TREE Lab
Up to the Phylogenetics main page
Goals
Today you will continue learning about maximum likelihood inference and will learn to use a program (IQ-TREE) known for the diversity of models it offers and its ability to handle large-scale phylogenetic analyses.
Objectives
After completing this lab, you will be able to…
- estimate a phylogeny using IQ-TREE
- obtain Ultrafast Bootstrap support values for the splits in the tree
- use IQ-TREE to find the optimal data partitioning scheme
- use BIC values to choose the best model (where model includes the tree as well as the substitution model)
- use IQ-TREE to find loci that conflict with other loci, suggesting paralogy, horizontal transfer, etc.
Getting started
I use a ![]() emoji below to indicate something you should do.
emoji below to indicate something you should do.
![]() Login to your account on the Storrs HPC cluster:
Login to your account on the Storrs HPC cluster:
ssh hpc
![]() Type the following to reserve an interactive session:
Type the following to reserve an interactive session:
gensrun
If that doesn’t work, it means you have not set up an alias for this task. You can type this each time you need an interactive session
srun -p general -q general --pty bash
or you can create an alias by opening the .bashrc file in your home directory:
nano ~/.bashrc
and typing the following (you can make it the first line of the file):
alias gensrun='srun -p general -q general --pty bash'
Save the .bashrc file and you can simply type gensrun to run the srun command the next time you login to the cluster!
![]() Create a directory named iqlab to serve as a playground for this week’s lab:
Create a directory named iqlab to serve as a playground for this week’s lab:
mkdir ~/iqlab
Create a text file in which to save answers
![]() Copy the text below into a text file to record you answers to questions posed in this lab (labeled with the
Copy the text below into a text file to record you answers to questions posed in this lab (labeled with the ![]() emoji).
emoji).
1. How many taxa are represented in the turtle.fa file?
answer:
2. How many loci are represented in the turtle.fa file?
answer:
3. How many substitution models were tested in the unpartitioned analysis?
answer:
4. What was the best model according to the Bayesian Information Criterion (BIC)?
answer:
5. Which of the 3 hypotheses (BC, BT, or CT) is supported by the ML tree for the unpartitioned analysis?
answer:
6. What is the BIC score for the unpartitioned ML tree?
answer:
7. What is the ultrafast bootstrap support for this hypothesis in the unpartitioned ML tree?
answer:
8. Which of the 3 hypotheses (BT, BC, or CT) is supported by the partitioned ML tree?
answer:
9. What is the ultrafast bootstrap support for this hypothesis in the partitioned ML tree?
answer:
10. What is the BIC score for the partitioned ML tree? Is it better (smaller) or worse (larger) than the BIC for the unpartitioned ML tree?
answer:
11. Is Tree1 the tree from the unpart or fullpart analysis?
answer:
12. Which 2 loci (counting over starting at 1 for the leftmost bar in the plot) stand out as saying something very different than the others?
answer:
13. What are these loci? (i.e. look them up in the turtles.nex file you downloaded at the start of the lab)
answer:
14. Which hypothesis of turtle relationships do these 2 loci support?
answer:
15. Which hypothesis of turtle relationships do 20 of the remaining 27 loci support?
answer:
16. Which hypothesis is now supported by each analysis and what is the ultrafast support and BIC score?
(fill out the table below):
---------------------------------------------------
Hypothesis ufboot BIC
---------------------------------------------------
unpart
---------------------------------------------------
fullpart
---------------------------------------------------
bestpart
---------------------------------------------------
17. Did merging partition subsets improve the fit of the model? What evidence did you use to decide this?
answer:
18. Did removing the paralogous loci improve confidence in one hypothesis over the others? What evidence did you use to decide this?
answer:
Download IQTREE
![]() Download the latest version (3.0.1) of IQ-TREE for “64-bit Linux Intel” (I got the link from the downloads web page):
Download the latest version (3.0.1) of IQ-TREE for “64-bit Linux Intel” (I got the link from the downloads web page):
curl -LO https://github.com/iqtree/iqtree3/releases/download/v3.0.1/iqtree-3.0.1-Linux-intel.tar.gz
The L is important in this case because there is a redirect involved (i.e. the url does not point directly to the file we need, so L tells curl to follow redirects until it gets to the final destination).
This should result in a file named iqtree-3.0.1-Linux-intel.tar.gz, which is a so-called “gzipped tar file”. The gz part of the name tells you that the file is compressed using the gzip algorithm, and the tar part of the name stands for Tape ARchive, which is an old term used for a single file that is actually a bunch of tiles concatenated together (for example, an entire directory of files). The tape part is because we used to use magnetic tape rather than hard drives to store computer data.
![]() How do you extract a
How do you extract a tar file? Using the tar command of course!
tar zxvf iqtree-3.0.1-Linux-intel.tar.gz
The zxvf options say to gunzip the file (z), then extract the files therein (x), be verbose (v) and the file to process follows (f). This will create a directory named iqtree-3.0.1-Linux-intel.
Navigate into that directory and then further navigate into the bin directory.
![]() Copy the
Copy the iqtree3 file (which is the actual program) to your own ~/bin directory so that it will be accessible from anywhere.
cd ~/iqlab
cd iqtree-3.0.1-Linux-intel/bin
mv iqtree3 ~/bin
![]() You can now delete the tar file and the directory that was created when we extracted it:
You can now delete the tar file and the directory that was created when we extracted it:
cd ~/iqlab
rm -rf iqtree-3.0.1-Linux-intel
rm -f iqtree-3.0.1-Linux-intel.tar.gz
(The -rf part tells the rm command to delete recursively - i.e. go into each subdirectory and get rid of everything, with the f part meaning “force”, which just tells rm to do its job without asking permission to delete every file it finds (which gets very tedious).
About IQ-TREE
IQ-TREE is known for its speed and ability to handle large nucleotide or protein datasets, and for the diversity of models it offers. Its main competitor is RAxML, which is also an excellent choice. We don’t have time to introduce you to every program that does phylogenetics; we’ve chosen IQ-TREE over RAxML mainly because it has truly excellent documentation. The explanation of options for this software is far better than for most other programs you will use this semester, and you can learn a lot about phylogenetics just looking up how to do things with IQ-TREE!
Start the tutorial
The tutorial below is a shortened version of the one written by Bui Minh (the primary developer of IQ-TREE) for the 2023 Woods Hole Workshop in Molecular Evolution.
Download the data
![]() Download the sequence data file (
Download the sequence data file (turtle.fa) and partition file (turtle.nex) that we will be analyzing today:
cd ~/iqlab
curl -O http://www.iqtree.org/workshop/data/turtle.fa
curl -O http://www.iqtree.org/workshop/data/turtle.nex
The file turtle.fa contains the actual sequence data. It is in FASTA format, which is very simple: the data for each taxon begins with a separate line beginning with a greater-than symbol (>) followed by the taxon name and sometimes other information. The sequence data for that taxon begins on the next line and continues over possibly several lines until the end of the file or another > is encountered.
How many taxa are represented in the turtle.fa file?
The file turtle.nex is a NEXUS format file but does not contain sequence data! Instead this file contains only a sets block with charset statements showing the range of sites corresponding to each locus.
Background
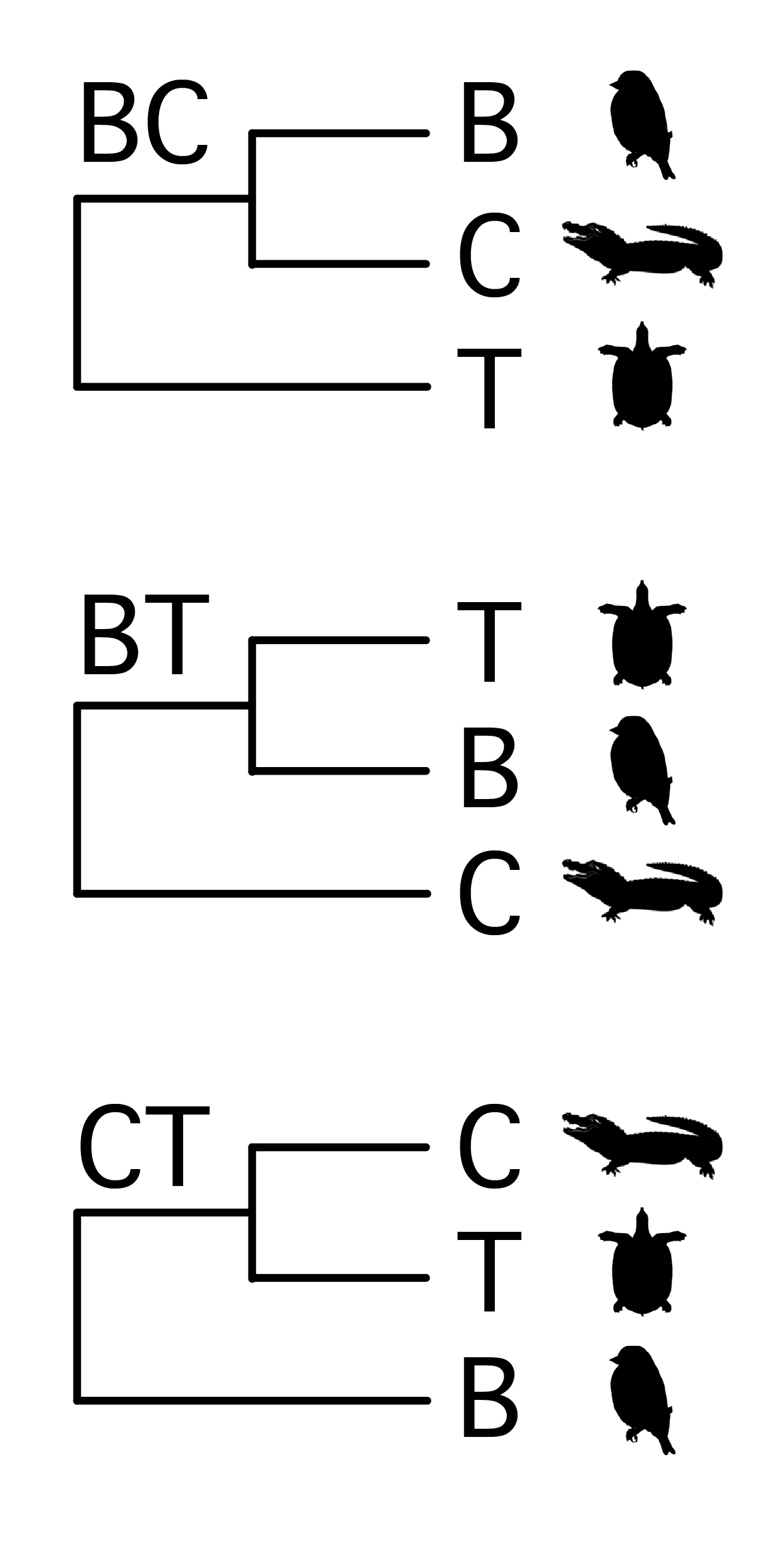
We will use these data to infer relationships between three familiar groups: turtles (T), crocodiles (C), and birds (B). There are three possible rooted trees for the three taxa, shown in the figure on the right (image credit: Phylopic). I will refer to these three hypotheses simply by the taxa that are sister to each other: i.e. BC, BT, CT.
These data represent a small subset of the 248-locus, 16-taxon data from Chiari et al. 2012, who concluded that their data “…unambiguously support turtles as a sister group to birds and crocodiles” (i.e. BC) however they found that CT was supported by “more simplistic” models and attributed this artifactual grouping to substitutional saturation at 3rd codon positions.
During the lab today, you will discover that a couple of loci tell a different story than the others and are responsible for the tendency to get the CT grouping under common models. The fact that these loci are paralogs was discovered by Jeremy Brown and Robert Thomson in 2017.
Let’s try to figure out which two loci are the paralogs.
Unpartitioned analysis
![]() Create a directory inside
Create a directory inside iqlab named unpart. Use the cd command to navigate into the unpart directory.
![]() Use this command to estimate the tree using maximum likelihood criterion:
Use this command to estimate the tree using maximum likelihood criterion:
iqtree3 -s ../turtle.fa -B 1000 -nt AUTO --prefix unpart
Here is a key to the settings we chose on the command line:
-
-sspecifies that the sequence data is in the fileturtle.fa -
-Bsays to perform 1000 ultrafast bootstraps -
-nt AUTOsays to figure out automatically how many threads to use -
--prefix unpartsays to begin the name of every output file withunpart
Note that we had to specify ../turtle.fa rather than turtle.fa because this file is located in the parent directory (iqlab) of the unpart directory in which you are now located.
A node in a cluster is a machine that may have multiple cores, where a core is a single processor. A thread is a part of a program that can be run independently of other parts. A single program can use multiple threads of execution to perform calculations in parallel, saving time. Ideally, each thread operates on a different core, otherwise threads compete with one another for time on the processor.
By specifying -nt AUTO (“nt = number of threads”), IQ-TREE will use as many cores as possible on the node that you were assigned using the srun command (and will use one thread per core). If you leave off the -nt specification, IQ-TREE will tell you in its output how many threads it could have used, but will actually just use 1 of them.
The -B setting tells IQ-TREE to assess support for individual edges (splits) using ultrafast bootstrapping. We have discussed what bootstrapping is in lecture (and how ultrafast bootstrapping differs from standard bootstrapping), but, for now, just know that this option will end up assigning a value to each edge in the summary consensus tree that tells you how plausible that edge is. Edges with ultrafast bootstraps > 95 are strongly supported, values smaller than that are more questionable.
This analysis will take a while (most of the time is spent figuring out which substitution model is best), so you might use the time to read on past the next couple of questions to get some background. Once the unpartitioned analysis finishes, view the output file unpart.iqtree in your favorite text editor (nano, BBEdit, Notepad++, etc.) and answer these questions:
An F in a model name means “empirical nucleotide frequencies”, an I in the name means “proportion of invariable sites”, a G means discrete Gamma among-site rate heterogeneity, and an R refers to the “free rates” model of among-site rate heterogeneity.
BIC will make more sense once you’ve had the introduction to Bayesian statistics, but, for now, just remember that models with smaller BIC values are better. BIC and the Akaike Information Criterion (AIC) are alternatives to using likelihood ratio tests and have the advantage that the models compared do not need to be nested. We will exclusively use BIC in this lab, but AIC would probably make the same model choices. The two approaches differ only in how much they penalize models for complexity (i.e. number of estimated parameters), with BIC imposing a larger penalty for each extra parameter.
Considering that crocodiles C includes alligator and caiman, birds B includes Taeniopygia and Gallus, and turtles T includes Emys, Chelonoidis, Caretta, and Phrynops…
You can answer the following question from looking at the text-based depiction of the tree in unpart.iqtree or you can download unpart.treefile to your laptop and open it in FigTree for a graphical depiction. To see the bootstrap support, type ufboot when it asks for label upon opening the file, then check the “Branch labels” check box, expand that tab, and specify ufboot for “Display”. When FigTree opens, if there is some information about nodes or edges that is encoded in the tree description, it asks you what you want to call it. I told you to call it ufboot but you could have typed something else. Whatever you type will show up in the “Display” dropdown control.
Partitioned analysis
Let’s now analyze the data when every locus has its own distinct relative rate. This means adding 28 relative rate parameters, one for each locus except for one (that final rate can be obtained from the others because the average relative rate must equal 1).
![]() Create a directory named
Create a directory named fullpart under iqlab that is at the same level as your unpart directory and navigate into the new fullpart directory.
![]() Enter this command to run IQ-TREE:
Enter this command to run IQ-TREE:
iqtree3 -s ../turtle.fa -p ../turtle.nex -B 1000 -nt AUTO --prefix fullpart
Some of these options you saw before, but here is a key to all of them anyway:
- the
-sspecifies the sequence data file - the
-pspecifies the partition file (which shows which sites are associated with each locus) - the
-Bspecifies the number of ultrafast bootstraps - the
-nt AUTOmeans “determine the number of threads to use automatically” with files created earlier - the
--prefix fullpartmeans that each output file will start withfullpart
Looking for outlier loci
Hopefully you discovered that partitioning makes a difference in which hypothesis of turtle relationships is favored. The question we’ll ask in this section is: “Do all gene loci agree, or is there conflict among loci as to the best topology”.
We’ll do this by computing the log likelihood for each locus on each of the two trees. The log likelihood is a measure of how well the model (which includes the tree topology and edge lengths) fits the data for a particular locus. If the log likelihood for a locus is higher on tree 1 than it is on tree 2, then the data for that locus fit tree 1 better (i.e. we are less surprised at the data for that locus if we assume tree 1 was the true tree).
![]() Create a directory named
Create a directory named locuscomp (short for “locus comparison”) at the same level as fullpart and unpart and navigate into it.
First, we need to get the two trees we want to compare into the same file.
![]() Use the
Use the cat (“concatenate”) command to merge the two files unpart.treefile and fullpart.treefile into a new file named turtle.trees:
cat ../unpart/unpart.treefile ../fullpart/fullpart.treefile > turtle.trees
![]() Second, run IQ-TREE to generate a file of locus-log-likelihoods:
Second, run IQ-TREE to generate a file of locus-log-likelihoods:
iqtree3 -s ../turtle.fa -p ../fullpart/fullpart.best_scheme.nex -z turtle.trees -n 0 -wpl --prefix locuscomp
Here is a key to the settings:
- the
-sspecifies the sequence data file - the
-pspecifies the partition file (we’ll use the partitioning scheme from thefullpartanalysis) - the
-zspecifies the file containing the trees to compare - the
-n 0means “do zero iterations to improve the likelihood” (we just want the likelihood of the tree as it is) - the
-wplinvokes the “writing partition log-likelihoods” functionality - the
--prefix locuscompmakes all output file names begin withlocuscompso that we don’t overwrite files created earlier
When the analysis is finished, you should find a file named locuscomp.partlh in the directory. Make a copy of this file so that we can modify it without destroying the original:
cp locuscomp.partlh locuscomp.R
The copy ends in the filenname extension .R. This is because we are going to convert this file (i.e. edit it in a text editor) into a file containing commands that can be executed by the programming language R.
Open the file locuscomp.R in a text editor (e.g. nano). It should look like this:
2 29
Tree1 -5088.86 -3413.96 -3626.17 -4298.08 -6232.66 ... -4394.17
Tree2 -5091.9 -3415.46 -3609.65 -4288.8 -6222.02 ... -4389.71
I’ve used elipses (...) to stand for 24 values left out. The 29 numbers on the 2nd and 3rd lines of this file are the log likelihoods for the loci specified in the ../fullpart/fullpart.best_scheme.nex file for one of the two trees.
![]() Delete the first line (
Delete the first line (2 29) of this file and edit the two remaining lines to look like this (except your version will not have any ellipses!):
Tree1 <- c(-5082.67, -3414.03, -3626.37, -4298.16, ..., -4394.24)
Tree2 <- c(-5086.25, -3415.55, -3609.85, -4288.83, ..., -4389.8)
(Note the commas separating each value - don’t forget to add those.)
When R reads these lines, it will make two vectors of values corresponding to the log likelihoods of the 29 loci for Tree1 and Tree2
![]() Now add a command at the end of the file to compute a vector of differences between the log likelihoods for Tree1 and Tree2 for each locus:
Now add a command at the end of the file to compute a vector of differences between the log likelihoods for Tree1 and Tree2 for each locus:
diffs <- Tree1 - Tree2
In R, subtracting one 29-element vector from another 29-element vector creates a new vector of length 29 containing the differences between each element.
![]() Finally, add one more line telling R to create a bar plot showing these 29 differences graphically:
Finally, add one more line telling R to create a bar plot showing these 29 differences graphically:
barplot(diffs)
Your final locuscomp.R file should now look like this (but, again, with no ellipses):
Tree1 <- c(-5082.67, -3414.03, -3626.37, -4298.16, ..., -4394.24)
Tree2 <- c(-5086.25, -3415.55, -3609.85, -4288.83, ..., -4389.8)
diffs <- Tree1 - Tree2
barplot(diffs)
![]() Load the R module so that you can use R to run this file:
Load the R module so that you can use R to run this file:
module load r
![]() Now execute your
Now execute your locuscomp.R file in R:
R --no-save < locuscomp.R
This should generate a file named Rplots.pdf.
![]() Pull this file back down to your laptop (there is no easy way to view it on the cluster) using Cyberduck or
Pull this file back down to your laptop (there is no easy way to view it on the cluster) using Cyberduck or scp and open it.
You should find that these two loci are the ones identified by Brown and Thomson 2019 as paralogs (see first paragraph of p. 522).
Excluding the outliers
Let’s rerun both the unpart and fullpart analyses without these 2 loci and see if they now agree on turtle relationships.
![]() Create a new directory
Create a new directory final at the same level as unpart, fullpart, and locuscomp and navigate into it.
![]() Copy the
Copy the turtle.nex file from the parent directory into your new final directory:
cp ../turtle.nex finalpart.nex
cp ../turtle.nex finalunpart.nex
![]() Edit the
Edit the finalpart.nex in nano, removing the two lines corresponding to the two paralogous loci.
![]() Edit the
Edit the finalunpart.nex file to remove the two problematic loci and pool sites from all other loci into a single big subset. Your finalunpart.nex file should look like this:
#nexus
begin sets;
charset allorthologs = 1-9822 10369-12795 13243-20820;
end;
![]() Run IQ-TREE to estimate an ML tree for the unpartitioned data:
Run IQ-TREE to estimate an ML tree for the unpartitioned data:
iqtree3 -s ../turtle.fa -p finalunpart.nex -B 1000 -nt AUTO --prefix unpart
![]() Run IQ-TREE to estimate an ML tree for the partitioned data:
Run IQ-TREE to estimate an ML tree for the partitioned data:
iqtree3 -s ../turtle.fa -p finalpart.nex -B 1000 -nt AUTO --prefix fullpart
![]() Finally, ask IQ-TREE to use the partition-finder algorithm to merge loci that are evolving at a rate that is similar enough that they can be modeled using the same relative rate:
Finally, ask IQ-TREE to use the partition-finder algorithm to merge loci that are evolving at a rate that is similar enough that they can be modeled using the same relative rate:
iqtree3 -s ../turtle.fa -p finalpart.nex -B 1000 -nt AUTO -m MFP+MERGE -rcluster 10 --prefix bestpart
The new options we haven’t seen before are:
- the
-m MFP+MERGEinvokes the relaxed-clustering partition-finder algorithm - the
-rcluster 10says to ony examine the top 10% of partition schemes
---------------------------------------------------
Hypothesis ufboot BIC
---------------------------------------------------
unpart
---------------------------------------------------
fullpart
---------------------------------------------------
bestpart
---------------------------------------------------